المتلازمة - ليس مرضاً بحد ذاته، ولكن مجموعة من الأعراض المرضية يظهر أغلبها أو بعضاً منها قي حالات مرضية معينة، وعادة ما تسمى بإسم الشخص الذي قام بنشر بحث علمي عنها كمجموعة أعراض، وأغلب المتلازمات نتيجة عيوب في الصبغيات - الكروموسومات أو المورثات - الجينات.
في هذا الجزء من الموقع سنحاول الكتابة عن بعض المتلازمات بطريقة بسيطة وسهلة، تجيب على كل الأسئلة التي يمكن أن يطرحها والدي الطفل ، كما أنها معلومات كافية للعاملين في مجال خدمة ذوي الإحتياجات الخاصة
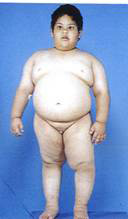
عرفت هذه المتلازمة عندما قام أخصائيو الغدد برادر – لابهارت – ويلي بنشر تقرير عام 1956 في المؤتمر الدولي الثامن لطب الأطفال في كوبنهاجن، عن حالات فيها صفات متعددة مثل : إعاقة عقلية ، الرخاوة، مشاكل في التغذية، السمنة، عدم اكتمال الاجهزة الجنسية، القصر، تاخر النمو، وغيرها، وفي الستينات من القرن الماضي تمت دراسات أخرى وتمت أضافة العديد من الاعراض، كما تمت أضافة الاعراض السلوكية في السبعينات، وقد أثبتت الدراسات أنه مع التحكم في النظام الغذائي فإن هذه الحالات يمكن أن تعيش لمدة أطول، وقد تم تتبع العديد من الحالات الذين بلغوا العشرينيات من العمر.
الانتشار :
o تعد متلازمة برادر ويلي من الاضطرابات الجينية النادرة إذ يقدر حدوثها حالة لكل 12.000-15.000 ولادة
o تتساوى نسبة الإصابة بين كل من الذكور والإناث
o كل الأعراق
o أغلب الحالات غير وراثية، ولكنها تحدث كطفرة جينية لأسباب غير معروفة
o في نسبة قليلة لا تتجاوز 2% وجد لها أسباب وراثية وترتفع نسبة تكرار الحالة لدى العائلة
o أحتمالية التكرار ضعيفة ماعدا الحالات النادرة وهي imprinting mutations, translocations, or inversions
الأسباب:
تنتج متلازمة برادر ويلي في معظم حالاتها من غياب أو حذف لجين من الذراع الطويل لكروموسوم 15 القادم من الأب ويرمز لها 15q11-q13، وفي بعض الأحيان تنتج من حصول الطفل المصاب على نسختين من الكروموسوم 15 من الأم، وهنا لا بد من توضيح الأمر :
o السبب في النقص في طرف الكروموسوم رقم 15 غير معروف
o وجد أن النقص يكون في الكروموسوم المنتقل من الأب بدون أسباب وراثية في 70% من الحالات
o في 25% من الحالات وجد أن هناك الزوج من الكروموسوم رقم 15 من الأم بدون وجود كروموسوم الأب
o في 2-5% من الحالات وجد أن النسخة رقم 15 من الأب في الكروموسوم لا تعمل.
.gif)
الخصائص والأعراض المرضية :
o نقص الحركة وهو جنين .
o ضعف العضلات منذ الطفولة ويزيد مع العمر
o صعوبة في الرضاعة
o خمول في مرحلة الطفولة مع بكاء ضعيف.
o تأخر عام في النمو الحركي والفكري
o ملامح جسمية مميزة
o نقص وعدم زيادة في الوزن في مرحلة الطفولة.
o شراهة في الأكل، بدانة بمرور الوقت، زيادة أو سرعة في الوزن مع سمنة مفرطة في غياب التدخل العلاجي .
o مشكلات في الأكل ، مثل الارتباط الزائد بطعام معين والأكل الزائد.
o تخلف عقلي بسيط أو صعوبات تعلم .
o القصور الجنسي والأعضاء التناسلية
o مشاكل سلوكية
o الوفاة المبكرة بسبب زيادة الوزن
.jpg)
.jpg)
الأعراض الجسمية :
o ملامح جسمية مميزة
o رأس صغير
o استطالة الجمجمة مع وجه ضيق وعينين ضيقتين وبيضاويتين
o أنخفاض الفم
o شعر أشقر وعيون زرقاء مهما كانت الجنسية في 50% من الحالات
o قصر القامة، قصر الأيدي والأقدام
.jpg)
.jpg)
ليونة العضلات :
من العلامات المميزة لمتلازمة برادر ولي، ويبدأ من الولادة ويتحسن تدريجياً مع التقدم في العمر، ومن علاماته في مرحلة الطفولة ضعف التغذية وعدم المقدرة على المص ، ضعف البكاء، ضعف الحركة وتأخر التطور الحركي، كما يلاحظ عجز في التنسيق الحركي، كسل العين – الحول.
.jpg)
العوق الفكري:
من العلامات المميزة غير معروفة السبب ، حيث لوحظ أن أكثر 95% من الحالات لديهم درجة من درجات التخلف الفكري، وفي الغالب يكون معدل الذكاء بين 70-79 ، ولكن يمكن أن يكون أقل من ذلك، أو يكون في المعدل الطبيعي.
الأطفال الذين لديهم معدل ذكاء طبيعي لوحظ لديهم صعوبات في التعلم، ضعف التركيز والإنتباه، ولكن لديهم نقاط قوة مثل الذاكرة ، القدرة على القراءة، وغيرها.
.jpg)
النطق والكلام :
ارتخاء العضلات قد يؤدي لصعوبات في التغذية، كما يؤدي إلى ضعف حركات الفم ومن ثم تأخر النطق والكلام، وهو ما قد يعني الاحتياج للتدريب على النطق والكلام ، وقد يحتاج الأمر إلى التواصل عن طريق لغة الإشارة، وعادة ما تتحسن اللغة لدى هؤلاء الأطفال، وتكون لغتهم جيدة.
ضعف النمو :
ضعف النمو يبدأ من الولادة، حيث يلاحظ ضعف الرضاعة، مما قد يستدعي التغذية عن طريق الأنبوب الفمي المعدي، وهنا يجب ملاحظة أحتياج الطفل من الغذاء كما الزيادة الطبيعية للوزن.
عادة ما يكون هرمون النمو ناقصاً لدى هؤلاء الأطفال، وهو ما يؤدي لقصر القامة، كما زيادة نسبة الدهون، ويحتاج الطفل إلى تعويض الهرمون الناقص في مرحلة الطفولة كما البالغين.
.jpg)
البدانة والشهية الشرهة:
الشراهة في الأكل سببها مركزي في الدماغ، لعدم عمل الأجزاء الخاصة بالشبع، قد لا تكون ملاحظة في السنوات الأولى من العمر لضعف عضلات المص والبلع، ولكنها تزداد مع التقدم في العمر.
عدم التحكم في كمية الطعام وضعف حركة الجسم تؤدي إلى السمنة، وتظهر البدانة عادة بعد سن الثانية أو الثالثة من العمر، لذى يجب أن يتناول المصابين كمية سعرات حرارية أقل من أقرانهم لمنع حدوث البدانة، وهو من الاسباب الرئيسة للوفاة، وحتى الآن لم يكتشف دواء قادر على ايقاف الشراهة في الأكل، وحتى الجراحة لا تجدي في تلك الحالات.
المشاكل السلوكية:
عادة ما يكون هؤلاء الأطفال طبيعيين هادئين في المراحل الأولى قبل سن المدرسة، ويحدث التغيير بعد ذلك، حيث يلاحظ عليهم تكرر حالات الغضب- الأنفعال- الأنطواء – الروتين، وتلك المشاكل غير مرتبطة بالسلوكيات الغذائية، ولكن التغيرات الغذائية يمكن أن تزيد حدتها، وعادة ما يجدي استخدام الأدوية النفسية في التخفيف من تلك السلوكيات.
.jpg)
القصور الجنسي:
من العلامات المميزة لمتلازمة برادر ولي، حيث يلاحظ :
o عدم أكتمال الأعضاء التناسلية الخارجية في الذكور، صغر حجم القضيب، تأخر نزول الخصيتين، وهو ما قد يحتاج للتدخل الجراحي
o عادة ما تبدأ التطورات الجنسية في عمر مبكر مثل ظهور شعر العانة، وتتوقف قبل الوصول إلى مرحلة البلوغ.
o تأخر نزول الطمث لبعد سن 16أو عدم نزوله لدى الإناث
o نقص الهرمونات الجنسية testosterone and estrogen
o التدخل العلاجي بالهرمونات ذي نتائج جيدة ولكن هناك ردود فعل سيئة من استخدامه
o عادة ما يكون المصابين غير قادرين على الإنجاب، ولكن سجل العديد من الحالات التي حصل بها حمل وانجاب.
.jpg)
التشخيص:
o الأعراض المرضية، وهناك قواعد معينة للتشخيص من حيث عدد العلامات الرئيسة والعلامات غير الرئيسة في كل مرحلة عمرية، وأخصائي الأمراض الوراثية لديهم هذه القواعد والجداول التابعة لها.
o DNA methylation analysis confirms diagnosis
o the specific genetic cause and associated recurrence risk FISH and DNA techniques can identify
.jpg)
.jpg)
.jpg)
هل يساعد التشخيص المبكر الأطفال المصابين بمتلازمة برادر ويلي ؟
في البداية لا بد من التذكير أن الحالة نتيجة عيوب في الكروموسومات وتلك لا يمكن علاجها أو تغييرها، ولكن التشخيص المبكر يعطي الوالدين الفرصة للتعرف على الحالة والتدرب على مواجهة التحديات المستقبلية، كما البحث عن طرق الدعم والمساعدة، فمثلاً معرفة وجود التأخر في النمو الحركي والفكري سيساعدهم في عدم البحث عن المسببات لهذا التأخر، ومعرفة الطرق للتعامل مع هذا التأخر، كما يمكن التعرف على العائلات ممن لديهم مثل هذه الحالة لمعرفة العقبات التي واجهتهم وطرق التعامل معها ومن أهمها طرق التعامل مع الشراهة في الأكل.
ما هو مستقبل الأطفال المصابين بمتلازمة برادر ويلي ؟
مع وجود الكثير من المشاكل الجسمية والسلوكية لدى المصابين بمتلازمة برادر ويلي، ولكن بمساعدة العائلة والمدرسة والمجتمع يمكن لهم الحياة في المجتمع والعمل بصورة أشبه بالطبيعية، ولكن ذلك يحتاج إلى الكثير من الجهد والوقت والتعاون بين جميع المؤسسات ذات العلاقة.
في الماضي الأطفال المصابين بمتلازمة برادر ويلي كانوا يتوفون في مرحلة مبكرة، وخاصة في مرحلة المراهقة نتيجة السمنة المفرطة، ولكن مع التحكم في السمنة والتغذية من خلال العلاج النفسي – الغذائي – الدوائي، أمكن لهؤلاء الأطفال من العيش لعمر أطول، والدراسات والأبحاث مستمرة للبحث عن طرق جديدة لمساعدتهم
.jpg)
مرض وراثي ينتقل من جيل لأخر عن طريق ما يسمى بالوراثة السائدة autosomal dominant trait، أول من قام بوصفها ونشرها الدكتور مارفان Dr. Antoine Marfan عام 1896م، وهي حالة تحدث نتيجة خلل في تركيبة النسيج الضام - Connective Tissue- والذي يدخل في تركيبة العديد من أعضاء الجسم، مما يؤدي إلى ظهور العديد من العيوب في العظام، الجهاز الدوري، العين، وغيرها، كما يؤدي إلى صعوبة في التعلم.
.jpg)
نسبة الانتشار والأسباب :o حالة نادرة تصيب فرد لكل 10.000
o يصيب الذكور والإناث بنفس النسبة
o تحدث الأعراض نتيجة لخلل في مورث- جين- يسمى بمورث الفيبريلين رقم واحد(Fibrillin 1) الموجود على الذراع الطويلة لكروموسوم chromosome 15q21.1.
o تدخل هذه المادة في تركيب النسيج الضام ، ومن ثم في تركيبة العديد من أعضاء الجسم كالأوعية الدموية، صمامات القلب،العظام، الجلد، والعين .
o مرض وراثي ينتقل من جيل لأخر عن طريق ما يسمى بالوراثة السائدة autosomal dominant trait
o اذا كان أحد الوالدين مصاب - هناك أحتمالية أنتقال الحالة لأطفاله 50%
o اذا كان أحد الوالدين مصاب، ولديهم طفل مصاب، فإن احتمالية تكرار الحالة 50% من المواليد
o في حوالي 25 بالمائة من الحالات يحدث لدى عائلة ليس فيها مصاب وهو ما يسمى بالطفرة الوراثية Mutation
الأعراض :
تختلف الأعراض المرضية بين شخص وآخر، كما تختلف بين أفراد العائلة الواحدة المصابين بالحالة، فقد نجد لدى البعض عرض أو أثنين، وقد تكون شديدة لدى الآخرين، ومن أهم الأعراض :
o طول القامة والأطراف
o الشكل المميز للرأس والوجه
o التغيرات في العمود الفقري والصدر
o مشاكل العين
o مشاكل في الجهاز الدوري والقلب
o كثرة حدوث الفتوق الاربية والفخذية
o خلوعٍ المفصل المتكرر
o تخلف عقلي بسيط في بعض الحالات
o صعوبة بالتعلم في بعض الحالات غير المصابة بتخلف عقلي
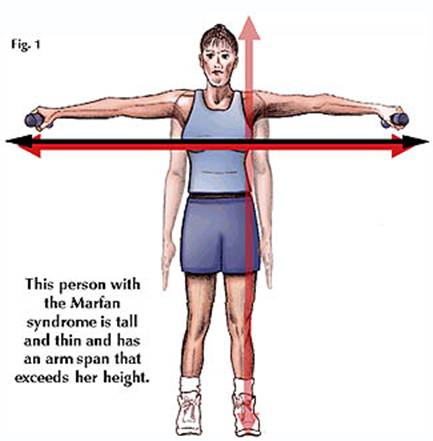
الشكل العام والأطراف :
o طول القامة نتيجة لطول عظام الأطراف - أطول من المعدل الطبيعي نسبة لعمرهم ولباقي أفراد أسرتهم
o الأطراف العليا والسفلي نحيفة و طويلة
o يكون قيـاس باعه - البعد بين الذراعين - أكبر من طول الجسم
o يزيد قياس القدم الى العانة عن البعد بين العانة وقمة الرأس
o طول ملحوظ في أصابع اليدين والقدمين
o تكون الأصابع طويلة دقيقة مفرطة البسط - عنكبية الأصابع Arachnodactyly
o علامة المعصم
o تكون عضلات الهيكل ضعيفة النمو وناقصة التوتر
o يؤدي ضعف الأربطة إلى الحركة المضاعفة للمفصل أو رخاوته
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
الرأس والوجه:o يبدو رأس طويل مع صغر حجم الوجه
o بروز عظام جبهة الرأس
o ارتفاع في سقف الحلق
o تكون الأسنان متزاحمة ومتراكبة
o يكون الحنك مرتفعاً ومتقوساً
العمود الفقري والصدرo الصدر الغاطس: عظمة القص - الصدر- غائر - صدر مقعر بشكل واضح، يجعل الصدر يأخذ شكل صدر الحمامة
.jpg)
o الصدر الناطح: عظمة القص - الصدر- بارزة إلى الأمام بشكل واضح - قمعي
.jpg)
o الظهر والعمود الفقري قد يزيد تقوسه - الحدب الجنفي
.jpg)
الجهاز الدوري- مشاكل القلب والشريان الأبهر :المشاكل القلبية وأصابات الشريان الأبهر - الأورطى - تؤثر في 80-90% من المصابين بمتلازمة مارفان، وهي السبب الرئيسي في أغلب حالات الوفاة، ومن أهم المشاكل:
o ارتخاء الصمام الميترالي
o ترجيع الصمام الميترالي
o توسع جذر الأبهر (الأورطي)
o ترجيع الصمام الأبهر
o توسع و تورم دموي في جذر الأبهر Aneurysm
.jpg)
.gif)
الرئتين :
تكون الألياف الضامة جدران الأكياس الهوائية في الرئة، ووجود الضعف فيها يجعلها غير قابلة للتمدد والانقباض، مما قد يؤدي إلى التمدد الزائد أو تكرار الانفجار، مما يؤدي إلى وجود هواء خارج الأكياس الرئوية pneumothorax
العلامات السلوكية والعقلية:
o تخلف عقلي بسيط
o صعوبة بالتعليم في بعض الحالات غير المصابة بتخلف عقلي
o المشاكل السلوكية والنفسية والاجتماعية المصاحبة
مشاكل العين :تظهر مشاكل العين في نصف المصابين تقريباً، فقد يؤدي ضعف الأنسجة الداعمة لعدسة العين إلى خلعها الجزئي، وغالباً للاتجاه نحو الأعلى ونحو الأمام، وهناك مشاكل في العين والنظر منها:
o تسطح القرنية
o قصر النضر
o ازرقاق في بياض العين
o عيوب العدسات
o أنفصال الشبكية
o الماء الأبيض - الساد
o الماء الأزرق - الجلوكوما
.jpg)
.jpg)
التشخيص :- ليس كل فرد طويل أو أطرافه طويلة مصاب بهذه المتلازمة، بل قد يكون الطول طبيعياً متوارثاً في العائلة
- ليس هناك تحليل في المختبرللتشخيص
- لذى فإن الطبيب المتخصص يمكنه الوصول للتشخيص من خلال ما أتفق عليه المتخصصون من قواعد للتشخيص المسماة Ghent criteria، وذلك بعد القيام بما يلي :
o تاريخ العائلة الطبي
o هل يوجد في العائلة أفراد طوال القامة
o هل يوجد في العائلة مصابون بالامراض القلبية
o الفحص الطبي السريري
o الكشف بالأشعة الصوتية
التشخيص الفارق والحالات المشابهة:
ليس كل حالات طول القامة هي حالات متلازمة مارفان، فهناك الحالات العائلية الطبيعية، كما أن هناك حالات أخرى منها:
o متلازمة أهلر - دانلوس Ehlers-Danlos Syndrome
o متلازمة الكروموسوم الجنسي الهش Fragile X Syndrome
o متلازمة كلاينفلتر Klinefelter Syndrome
o مرض الضخامة Gigantism and Acromegaly
o ارتفاع هرمون الغدة النخامية Hyperpituitarism
o ارتفاع هرمون الغدة الدرقية Hyperthyroidism
العلاج والعناية العامة :o عناية صحية مستمرة
o المتابعة الدورية مع طبيب مختص في أمراض القلب، وقد يحتاج الأمر إلى إجراء الأشعة الصوتية للقلب كل ستة أشهر
o عناية تعليمية خاصة للمتخلفين عقليا ومن لديهم صعوبات تعلم
o عناية خاصة بالعين ومشاكل رؤية
o الجراحة : قد تحتاج بعض الحالات إلى التدخل الجراحي المبكر مثل حالات توسع الشريان الأبهر وغيره
o تجنب الأنشطة الرياضية التي يصاحبها أحتكاك وتصادم ، مما يمكن أن تؤديه من مشاكل قلبية أو أضرار بالعين
ما هو مآل الحالة ؟متلازمة مارفان مستمرة طوال العمر، تختلف الأعراض لكل مرحلة عمرية، كما تختلف من شخص لآخر حسب درجة تأثيراتها، ومع المتابعة المستمرة، والتدخل العلاجي والجراحي فإن النتائج جيدة.
في هذا الجزء من الموقع سنحاول الكتابة عن بعض المتلازمات بطريقة بسيطة وسهلة، تجيب على كل الأسئلة التي يمكن أن يطرحها والدي الطفل ، كما أنها معلومات كافية للعاملين في مجال خدمة ذوي الإحتياجات الخاصة
متلازمة برادر- ويلي
Prader-Willi syndrome
عرفت هذه المتلازمة عندما قام أخصائيو الغدد برادر – لابهارت – ويلي بنشر تقرير عام 1956 في المؤتمر الدولي الثامن لطب الأطفال في كوبنهاجن، عن حالات فيها صفات متعددة مثل : إعاقة عقلية ، الرخاوة، مشاكل في التغذية، السمنة، عدم اكتمال الاجهزة الجنسية، القصر، تاخر النمو، وغيرها، وفي الستينات من القرن الماضي تمت دراسات أخرى وتمت أضافة العديد من الاعراض، كما تمت أضافة الاعراض السلوكية في السبعينات، وقد أثبتت الدراسات أنه مع التحكم في النظام الغذائي فإن هذه الحالات يمكن أن تعيش لمدة أطول، وقد تم تتبع العديد من الحالات الذين بلغوا العشرينيات من العمر.
الانتشار :
o تعد متلازمة برادر ويلي من الاضطرابات الجينية النادرة إذ يقدر حدوثها حالة لكل 12.000-15.000 ولادة
o تتساوى نسبة الإصابة بين كل من الذكور والإناث
o كل الأعراق
o أغلب الحالات غير وراثية، ولكنها تحدث كطفرة جينية لأسباب غير معروفة
o في نسبة قليلة لا تتجاوز 2% وجد لها أسباب وراثية وترتفع نسبة تكرار الحالة لدى العائلة
o أحتمالية التكرار ضعيفة ماعدا الحالات النادرة وهي imprinting mutations, translocations, or inversions
الأسباب:
تنتج متلازمة برادر ويلي في معظم حالاتها من غياب أو حذف لجين من الذراع الطويل لكروموسوم 15 القادم من الأب ويرمز لها 15q11-q13، وفي بعض الأحيان تنتج من حصول الطفل المصاب على نسختين من الكروموسوم 15 من الأم، وهنا لا بد من توضيح الأمر :
o السبب في النقص في طرف الكروموسوم رقم 15 غير معروف
o وجد أن النقص يكون في الكروموسوم المنتقل من الأب بدون أسباب وراثية في 70% من الحالات
o في 25% من الحالات وجد أن هناك الزوج من الكروموسوم رقم 15 من الأم بدون وجود كروموسوم الأب
o في 2-5% من الحالات وجد أن النسخة رقم 15 من الأب في الكروموسوم لا تعمل.
الخصائص والأعراض المرضية :
o نقص الحركة وهو جنين .
o ضعف العضلات منذ الطفولة ويزيد مع العمر
o صعوبة في الرضاعة
o خمول في مرحلة الطفولة مع بكاء ضعيف.
o تأخر عام في النمو الحركي والفكري
o ملامح جسمية مميزة
o نقص وعدم زيادة في الوزن في مرحلة الطفولة.
o شراهة في الأكل، بدانة بمرور الوقت، زيادة أو سرعة في الوزن مع سمنة مفرطة في غياب التدخل العلاجي .
o مشكلات في الأكل ، مثل الارتباط الزائد بطعام معين والأكل الزائد.
o تخلف عقلي بسيط أو صعوبات تعلم .
o القصور الجنسي والأعضاء التناسلية
o مشاكل سلوكية
o الوفاة المبكرة بسبب زيادة الوزن
الأعراض الجسمية :
o ملامح جسمية مميزة
o رأس صغير
o استطالة الجمجمة مع وجه ضيق وعينين ضيقتين وبيضاويتين
o أنخفاض الفم
o شعر أشقر وعيون زرقاء مهما كانت الجنسية في 50% من الحالات
o قصر القامة، قصر الأيدي والأقدام
ليونة العضلات :
من العلامات المميزة لمتلازمة برادر ولي، ويبدأ من الولادة ويتحسن تدريجياً مع التقدم في العمر، ومن علاماته في مرحلة الطفولة ضعف التغذية وعدم المقدرة على المص ، ضعف البكاء، ضعف الحركة وتأخر التطور الحركي، كما يلاحظ عجز في التنسيق الحركي، كسل العين – الحول.
العوق الفكري:
من العلامات المميزة غير معروفة السبب ، حيث لوحظ أن أكثر 95% من الحالات لديهم درجة من درجات التخلف الفكري، وفي الغالب يكون معدل الذكاء بين 70-79 ، ولكن يمكن أن يكون أقل من ذلك، أو يكون في المعدل الطبيعي.
الأطفال الذين لديهم معدل ذكاء طبيعي لوحظ لديهم صعوبات في التعلم، ضعف التركيز والإنتباه، ولكن لديهم نقاط قوة مثل الذاكرة ، القدرة على القراءة، وغيرها.
النطق والكلام :
ارتخاء العضلات قد يؤدي لصعوبات في التغذية، كما يؤدي إلى ضعف حركات الفم ومن ثم تأخر النطق والكلام، وهو ما قد يعني الاحتياج للتدريب على النطق والكلام ، وقد يحتاج الأمر إلى التواصل عن طريق لغة الإشارة، وعادة ما تتحسن اللغة لدى هؤلاء الأطفال، وتكون لغتهم جيدة.
ضعف النمو :
ضعف النمو يبدأ من الولادة، حيث يلاحظ ضعف الرضاعة، مما قد يستدعي التغذية عن طريق الأنبوب الفمي المعدي، وهنا يجب ملاحظة أحتياج الطفل من الغذاء كما الزيادة الطبيعية للوزن.
عادة ما يكون هرمون النمو ناقصاً لدى هؤلاء الأطفال، وهو ما يؤدي لقصر القامة، كما زيادة نسبة الدهون، ويحتاج الطفل إلى تعويض الهرمون الناقص في مرحلة الطفولة كما البالغين.
البدانة والشهية الشرهة:
الشراهة في الأكل سببها مركزي في الدماغ، لعدم عمل الأجزاء الخاصة بالشبع، قد لا تكون ملاحظة في السنوات الأولى من العمر لضعف عضلات المص والبلع، ولكنها تزداد مع التقدم في العمر.
عدم التحكم في كمية الطعام وضعف حركة الجسم تؤدي إلى السمنة، وتظهر البدانة عادة بعد سن الثانية أو الثالثة من العمر، لذى يجب أن يتناول المصابين كمية سعرات حرارية أقل من أقرانهم لمنع حدوث البدانة، وهو من الاسباب الرئيسة للوفاة، وحتى الآن لم يكتشف دواء قادر على ايقاف الشراهة في الأكل، وحتى الجراحة لا تجدي في تلك الحالات.
المشاكل السلوكية:
عادة ما يكون هؤلاء الأطفال طبيعيين هادئين في المراحل الأولى قبل سن المدرسة، ويحدث التغيير بعد ذلك، حيث يلاحظ عليهم تكرر حالات الغضب- الأنفعال- الأنطواء – الروتين، وتلك المشاكل غير مرتبطة بالسلوكيات الغذائية، ولكن التغيرات الغذائية يمكن أن تزيد حدتها، وعادة ما يجدي استخدام الأدوية النفسية في التخفيف من تلك السلوكيات.
القصور الجنسي:
من العلامات المميزة لمتلازمة برادر ولي، حيث يلاحظ :
o عدم أكتمال الأعضاء التناسلية الخارجية في الذكور، صغر حجم القضيب، تأخر نزول الخصيتين، وهو ما قد يحتاج للتدخل الجراحي
o عادة ما تبدأ التطورات الجنسية في عمر مبكر مثل ظهور شعر العانة، وتتوقف قبل الوصول إلى مرحلة البلوغ.
o تأخر نزول الطمث لبعد سن 16أو عدم نزوله لدى الإناث
o نقص الهرمونات الجنسية testosterone and estrogen
o التدخل العلاجي بالهرمونات ذي نتائج جيدة ولكن هناك ردود فعل سيئة من استخدامه
o عادة ما يكون المصابين غير قادرين على الإنجاب، ولكن سجل العديد من الحالات التي حصل بها حمل وانجاب.
التشخيص:
o الأعراض المرضية، وهناك قواعد معينة للتشخيص من حيث عدد العلامات الرئيسة والعلامات غير الرئيسة في كل مرحلة عمرية، وأخصائي الأمراض الوراثية لديهم هذه القواعد والجداول التابعة لها.
o DNA methylation analysis confirms diagnosis
o the specific genetic cause and associated recurrence risk FISH and DNA techniques can identify
هل يساعد التشخيص المبكر الأطفال المصابين بمتلازمة برادر ويلي ؟
في البداية لا بد من التذكير أن الحالة نتيجة عيوب في الكروموسومات وتلك لا يمكن علاجها أو تغييرها، ولكن التشخيص المبكر يعطي الوالدين الفرصة للتعرف على الحالة والتدرب على مواجهة التحديات المستقبلية، كما البحث عن طرق الدعم والمساعدة، فمثلاً معرفة وجود التأخر في النمو الحركي والفكري سيساعدهم في عدم البحث عن المسببات لهذا التأخر، ومعرفة الطرق للتعامل مع هذا التأخر، كما يمكن التعرف على العائلات ممن لديهم مثل هذه الحالة لمعرفة العقبات التي واجهتهم وطرق التعامل معها ومن أهمها طرق التعامل مع الشراهة في الأكل.
ما هو مستقبل الأطفال المصابين بمتلازمة برادر ويلي ؟
مع وجود الكثير من المشاكل الجسمية والسلوكية لدى المصابين بمتلازمة برادر ويلي، ولكن بمساعدة العائلة والمدرسة والمجتمع يمكن لهم الحياة في المجتمع والعمل بصورة أشبه بالطبيعية، ولكن ذلك يحتاج إلى الكثير من الجهد والوقت والتعاون بين جميع المؤسسات ذات العلاقة.
في الماضي الأطفال المصابين بمتلازمة برادر ويلي كانوا يتوفون في مرحلة مبكرة، وخاصة في مرحلة المراهقة نتيجة السمنة المفرطة، ولكن مع التحكم في السمنة والتغذية من خلال العلاج النفسي – الغذائي – الدوائي، أمكن لهؤلاء الأطفال من العيش لعمر أطول، والدراسات والأبحاث مستمرة للبحث عن طرق جديدة لمساعدتهم
متلازمة مارافانMarfan's Syndrome
نسبة الانتشار والأسباب :o حالة نادرة تصيب فرد لكل 10.000
o يصيب الذكور والإناث بنفس النسبة
o تحدث الأعراض نتيجة لخلل في مورث- جين- يسمى بمورث الفيبريلين رقم واحد(Fibrillin 1) الموجود على الذراع الطويلة لكروموسوم chromosome 15q21.1.
o تدخل هذه المادة في تركيب النسيج الضام ، ومن ثم في تركيبة العديد من أعضاء الجسم كالأوعية الدموية، صمامات القلب،العظام، الجلد، والعين .
o مرض وراثي ينتقل من جيل لأخر عن طريق ما يسمى بالوراثة السائدة autosomal dominant trait
o اذا كان أحد الوالدين مصاب - هناك أحتمالية أنتقال الحالة لأطفاله 50%
o اذا كان أحد الوالدين مصاب، ولديهم طفل مصاب، فإن احتمالية تكرار الحالة 50% من المواليد
o في حوالي 25 بالمائة من الحالات يحدث لدى عائلة ليس فيها مصاب وهو ما يسمى بالطفرة الوراثية Mutation
الأعراض :
تختلف الأعراض المرضية بين شخص وآخر، كما تختلف بين أفراد العائلة الواحدة المصابين بالحالة، فقد نجد لدى البعض عرض أو أثنين، وقد تكون شديدة لدى الآخرين، ومن أهم الأعراض :
o طول القامة والأطراف
o الشكل المميز للرأس والوجه
o التغيرات في العمود الفقري والصدر
o مشاكل العين
o مشاكل في الجهاز الدوري والقلب
o كثرة حدوث الفتوق الاربية والفخذية
o خلوعٍ المفصل المتكرر
o تخلف عقلي بسيط في بعض الحالات
o صعوبة بالتعلم في بعض الحالات غير المصابة بتخلف عقلي
الشكل العام والأطراف :
o طول القامة نتيجة لطول عظام الأطراف - أطول من المعدل الطبيعي نسبة لعمرهم ولباقي أفراد أسرتهم
o الأطراف العليا والسفلي نحيفة و طويلة
o يكون قيـاس باعه - البعد بين الذراعين - أكبر من طول الجسم
o يزيد قياس القدم الى العانة عن البعد بين العانة وقمة الرأس
o طول ملحوظ في أصابع اليدين والقدمين
o تكون الأصابع طويلة دقيقة مفرطة البسط - عنكبية الأصابع Arachnodactyly
o علامة المعصم
o تكون عضلات الهيكل ضعيفة النمو وناقصة التوتر
o يؤدي ضعف الأربطة إلى الحركة المضاعفة للمفصل أو رخاوته
الرأس والوجه:o يبدو رأس طويل مع صغر حجم الوجه
o بروز عظام جبهة الرأس
o ارتفاع في سقف الحلق
o تكون الأسنان متزاحمة ومتراكبة
o يكون الحنك مرتفعاً ومتقوساً
العمود الفقري والصدرo الصدر الغاطس: عظمة القص - الصدر- غائر - صدر مقعر بشكل واضح، يجعل الصدر يأخذ شكل صدر الحمامة
o الصدر الناطح: عظمة القص - الصدر- بارزة إلى الأمام بشكل واضح - قمعي
o الظهر والعمود الفقري قد يزيد تقوسه - الحدب الجنفي
الجهاز الدوري- مشاكل القلب والشريان الأبهر :المشاكل القلبية وأصابات الشريان الأبهر - الأورطى - تؤثر في 80-90% من المصابين بمتلازمة مارفان، وهي السبب الرئيسي في أغلب حالات الوفاة، ومن أهم المشاكل:
o ارتخاء الصمام الميترالي
o ترجيع الصمام الميترالي
o توسع جذر الأبهر (الأورطي)
o ترجيع الصمام الأبهر
o توسع و تورم دموي في جذر الأبهر Aneurysm
الرئتين :
تكون الألياف الضامة جدران الأكياس الهوائية في الرئة، ووجود الضعف فيها يجعلها غير قابلة للتمدد والانقباض، مما قد يؤدي إلى التمدد الزائد أو تكرار الانفجار، مما يؤدي إلى وجود هواء خارج الأكياس الرئوية pneumothorax
العلامات السلوكية والعقلية:
o تخلف عقلي بسيط
o صعوبة بالتعليم في بعض الحالات غير المصابة بتخلف عقلي
o المشاكل السلوكية والنفسية والاجتماعية المصاحبة
مشاكل العين :تظهر مشاكل العين في نصف المصابين تقريباً، فقد يؤدي ضعف الأنسجة الداعمة لعدسة العين إلى خلعها الجزئي، وغالباً للاتجاه نحو الأعلى ونحو الأمام، وهناك مشاكل في العين والنظر منها:
o تسطح القرنية
o قصر النضر
o ازرقاق في بياض العين
o عيوب العدسات
o أنفصال الشبكية
o الماء الأبيض - الساد
o الماء الأزرق - الجلوكوما
التشخيص :- ليس كل فرد طويل أو أطرافه طويلة مصاب بهذه المتلازمة، بل قد يكون الطول طبيعياً متوارثاً في العائلة
- ليس هناك تحليل في المختبرللتشخيص
- لذى فإن الطبيب المتخصص يمكنه الوصول للتشخيص من خلال ما أتفق عليه المتخصصون من قواعد للتشخيص المسماة Ghent criteria، وذلك بعد القيام بما يلي :
o تاريخ العائلة الطبي
o هل يوجد في العائلة أفراد طوال القامة
o هل يوجد في العائلة مصابون بالامراض القلبية
o الفحص الطبي السريري
o الكشف بالأشعة الصوتية
التشخيص الفارق والحالات المشابهة:
ليس كل حالات طول القامة هي حالات متلازمة مارفان، فهناك الحالات العائلية الطبيعية، كما أن هناك حالات أخرى منها:
o متلازمة أهلر - دانلوس Ehlers-Danlos Syndrome
o متلازمة الكروموسوم الجنسي الهش Fragile X Syndrome
o متلازمة كلاينفلتر Klinefelter Syndrome
o مرض الضخامة Gigantism and Acromegaly
o ارتفاع هرمون الغدة النخامية Hyperpituitarism
o ارتفاع هرمون الغدة الدرقية Hyperthyroidism
العلاج والعناية العامة :o عناية صحية مستمرة
o المتابعة الدورية مع طبيب مختص في أمراض القلب، وقد يحتاج الأمر إلى إجراء الأشعة الصوتية للقلب كل ستة أشهر
o عناية تعليمية خاصة للمتخلفين عقليا ومن لديهم صعوبات تعلم
o عناية خاصة بالعين ومشاكل رؤية
o الجراحة : قد تحتاج بعض الحالات إلى التدخل الجراحي المبكر مثل حالات توسع الشريان الأبهر وغيره
o تجنب الأنشطة الرياضية التي يصاحبها أحتكاك وتصادم ، مما يمكن أن تؤديه من مشاكل قلبية أو أضرار بالعين
ما هو مآل الحالة ؟متلازمة مارفان مستمرة طوال العمر، تختلف الأعراض لكل مرحلة عمرية، كما تختلف من شخص لآخر حسب درجة تأثيراتها، ومع المتابعة المستمرة، والتدخل العلاجي والجراحي فإن النتائج جيدة.
ليست هناك تعليقات:
إرسال تعليق